
Abstract
In this paper, we first review different strategies reported in the literature to elaborate electrochemical DNA sensors based on carbon nanotubes. Then we report a new strategy to graft both redox and DNA probes onto carbon nanotubes to make a label-free DNA sensor. Oxidized single-walled carbon nanotubes are first immobilized on a self-assembled monolayer of cysteamine. Then a redox probe, a quinone derivative 3-[(2-aminoethyl) sulfanyl-5-hydroxy-1,4-naphthoquinone], is grafted onto the free carboxylic groups of the nanotubes. After that, for DNA probe grafting, new carboxylic sites are generated via an aryl diazonium route. After hybridization with a complementary sequence, the conformational changes of DNA could influence the redox kinetics of quinone, leading to a current increase in the redox signal, detected by square wave voltammetry. The system is selective, as it can distinguish a single mismatched sequence from the complementary one.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
In recent years, electrochemical deoxyribonucleic acid (DNA) biosensors have been under intense investigation and they have recently been reviewed [1–5]. Compared with traditional optical methods, electrochemical DNA biosensors have a great advantage for rapid and inexpensive detection of specific DNA sequences. The principle of the system is to transform the hybridization of a DNA probe immobilized on the electrode to a visible change in electrochemical signal. Among various materials employed to enhance the transduction, carbon nanotubes (CNTs) are very attractive due to their high electrical conductivity, great specific surface area and excellent chemical stability.
CNTs have been one of the most promising nano-materials since their discovery by Iijima in 1991 [6]. They belong to the fullerene structural family and can be thought of as rolled up graphene sheets. Single-wall carbon nanotubes (SWCNTs) consist of a single carbon cylinder, whereas multi-wall carbon nanotubes (MWCNTs) are made of multiple walls. CNTs can be synthesized by an arc-discharge method, a laser-furnace method or chemical-vapor deposition (CVD). Their excellent electrical property suggests that CNTs have the ability to promote electron transfer reactions when used as electrodes in electrochemical reactions, which makes them a very good platform for electrochemical biosensors.
This article will discuss some advances in CNT application to the electrochemical detection of DNA. As DNA detection systems based on CNTs should imply CNT immobilization and DNA probe grafting, we will first discuss CNT immobilization methods, then DNA probe grafting methods, before presenting various electrochemical DNA sensor structures.
2. CNT immobilization methods
Among many different methods developed to immobilize CNTs on the electrode surface, the simplest and most direct is to put a dispersion of CNTs or functionalized CNTs on an electrode surface and then wait several hours to let it dry [7–9]. The CNTs will be immobilized on the surface by physisorption mode. With this principle, Lee et al went further to obtain an ultrathin functionalized MWCNT film via layer-by-layer assembly [10]. To summarize this technique briefly, first, negatively and positively charged MWCNTs were prepared, then two dispersions of these were made in distilled water. The substrates were dipped first into a positively charged MWCNT dispersion, washed in water and then dipped into negatively charged MWCNTs. This process was repeated to obtain the desired number of bilayers of MWCNT thin film. This system shows high electronic conductivity and its capacity can be precisely controlled.
Another simple approach is adding CNTs in the process of electropolymerization. Normally, functionalized CNTs were added to the solution of a monomer, and a conductive polymer film containing CNTs could be obtained on the electrode surface by electropolymerization [11–13]. By this means, the arrangement of CNTs could not be well controlled; however, the CNTs could obviously increase the specific surface of the electrode, reinforce the electron transfer and carry several functional groups, as carboxylic acids.
For a lot of applications, well-defined CNT arrays are highly desirable. Compared with previous methods, this model needs better control of CNT immobilization. Of course, a physical method, like CVD, can produce CNT arrays on the surface of the substrate directly. As for the chemical immobilization method, the technique of self-assembled CNTs through a linker of alkylthiol offers a chance of obtaining a monolayer of CNT arrays. The original idea of this approach came from the work of Liu et al [14]. They immersed a gold surface into a solution of thiol-functionalized CNTs and found a monolayer of CNTs was generated, thanks to the bonds that are easily formed between gold and thiol. Derivatives of this approach became very popular for CNT immobilization. Gooding and others have done excellent work on a variant of this approach. A self-assembled monolayer (SAM) of amine-terminated alkanethiol was first generated on a gold surface, and then oxidized SWCNTs which bring carboxylic acid moieties at the end and on the surface were immobilized on this monolayer through the amidation reaction between aminated distal end of the SAM and –COOH of SWCNTs [15–18]. They obtained vertically aligned SWCNT bundles on the SAM-modified gold surface from this approach and other molecules (redox group, enzyme, etc) could be grafted on the free ends of SWCNTs, which still have some carboxylic acid moieties. The influence of the chain length of alkanethiol to electron transfer was also investigated for this system [19]. The results suggest that electron transfer proceeds via tunneling through the SAM rather than SWCNTs disrupting the SAM. Kim and Lee [20] developed another derivative of this approach to obtain well-patterned SWCNT arrays by combining lithography and CNT self-assembly. A gold surface was first of all patterned by silica nanospheres, then a cysteamine SAM was grafted onto the free surface; acid chloride-functionalized SWCNTs were grafted onto the amine-modified distal end of the SAM by electrophoretic deposition. Similar vertically aligned SWCNT arrays were also fabricated on a silicon surface via a thioester linker instead of an amine-terminated alkanethiol [21].
3. DNA probe grafting methods
CNTs have been studied as potential carriers of various bio-components. To make electrochemical DNA sensors based on CNTs, DNA probe grafting has been paid great attention and considered as a fundamental methodology. A multistep route to covalently link DNA on SWCNTs has been reported [22]. SWCNT, have been treated first in acid to generate carboxylic moieties on the ends as well as on the walls of nanotubes, and then a bifunctional linker has been attached. A chemical reaction took place between the linker and thiol-terminated DNA strands. The resulting composites were found to hybridize selectively with the complementary sequences of oligonucleotides. A similar and more popular strategy is to link directly amine-terminated DNA to oxidized CNTs by diimide activation of the carboxylic moieties [23–25]. Alternatively, peptide nucleic acid could be grafted to activate the carboxylic moieties of CNTs, and then DNA could be attached to CNTs by hybridizing with grafted PNA [26].
Moghaddam et al [27] have reported an interesting DNA binding on MWCNT method using azide photochemistry. Aligned MWCNTs on the substrate were functionalized by the photoactive molecule azidothymidine (AZT) under UV light, then DNA was synthesized in situ from the sidewalls of CNTs. Finally they could achieve MWCNTs decorated with DNA with the desired sequences.
4. Structures of an electrochemical DNA sensor based on CNTs
Electrochemical DNA sensors rely on the immobilization of the DNA probe onto the transducer surface and the translation of the probe/target hybridization into a change in current (i.e. the transduction). Generally speaking, CNTs serve in this system as a part of the transducer that carries DNA probes and, at the same time, promotes electron transfer. The excellent sensitivity of CNT conductivity to the chemical environment changes around their surface and allows the use of CNTs as highly sensitive 'nanoscaled' sensors.
A DNA electrochemical sensor based on self-assembled MWCNTs has been described by Wang et al [28]. As the classic technique, self-assembled MWCNTs on an Au surface were treated by HNO 3 to introduce carboxylic acids on the nanotubes, and then amine-terminated ODN probes were grafted on MWCNTs by using diimide, which activates the amidation reaction. Hybridization between probe and target DNA was monitored using methylene blue as a redox indicator, which has a strong affinity for free guanine bases [29, 30]. Transduction is 'signal-off' type, i.e. redox currents drop down upon hybridization. A similar system with high selectivity and high sensibility has been reported by He [31]. In this system, self-assembled CNTs on an Au surface were functionalized by plasma to produce carboxylic moieties, and then amine-terminated ODN probes were grafted onto CNTs. Hybridization took place between unmodified ODN probes and a ferrocene-labeled DNA target, so that the signal of ferrocene was used to monitor hybridization. This system generates a 'signal-on' response, i.e. redox currents increase upon hybridization; however, it needs DNA labeling by ferrocene.
Vertically aligned MWCNTs embedded in SiO 2 have been used for ultrasensitive DNA detection [32]. In this configuration, oligonucleotide probes were also grafted to the open ends of nanotubes, but a better sensitivity has been obtained as sub-attomole DNA targets can be detected by combining these MWCNT arrays with Ru(bpy)3 2+-mediated guanine oxidation. This system shows that sensitivity is dramatically improved by lowering the nanotube density. Another interesting DNA detection system based on layer-by-layer MWCNTs has been described by Ma et al, which is constructed with a multistep surface functionalization [33]. MWCNTs activated on their two ends were covalently and vertically immobilized on a gold surface, then gold nanoparticles were grafted onto the free ends of MWCNTs. After that, new activated MWCNTs were immobilized on gold nanoparticles. The process was repeated several times to achieve the layer-by-layer system. The system was characterized by cyclic voltammetry and electrochemical impedance spectroscopy. Finally, amine-ended ODN probes were grafted onto the free ends of MWCNTs and hybridization was monitored by differential pulse voltammetry using doxorubicin as a redox indicator. Benefiting from the ability of CNTs to promote electron transfer and the catalytic activity of gold nano-particles, the sensibility of this DNA sensor was improved and good stability and reproducibility were obtained.
In this paper, we report a new CNT-using strategy for such a system. Simply, oxidized single-walled CNTs (SWNT-COOH) are first immobilized on a self-assembled monolayer of cysteamine; and then redox probes, a quinone derivative (3-[(2-aminoethyl)sulfanyl-5-hydroxy -1,4-naphthoquinone], called Jug-NH 2), are grafted onto the free carboxylic acid groups of SWNTs. After that, for DNA probe grafting, we generate new reactive sites (new carboxylic acids) via the electrochemical reduction of aryl diazonium salt. This technique has been successfully applied to the modification of a carbon electrode surface [34] and investigated for the modification of SWNTs [35, 36]. Finally, a DNA probe was grafted onto the new carboxylic acids and hybridization was studied with different DNA targets. The system construction is ideally and schematically illustrated in figure 1. As shown, the distribution of DNA and redox probes over the electrode surface is controlled by nanostructuration induced by SWNTs, which brings both redox and DNA probes together on a very small space (ideally, the end of each SWCNT). This improves the non-covalent interactions between them, so that redox current variations induced by the conformational change of the DNA probe could be improved as well. After hybridization, the change in physical properties of DNA probes (mostly conformational changes) could influence the redox kinetics of the quinone group and the conductivity of the SWNTs, which leads to a current increase in the redox signal. This results in a reagentless electrochemical transduction and 'signal on' system where SWNTs work as a nanodomain onto which two kinds of probes (the redox transducer and the biomolecule) are grafted and forced to interact with each other.
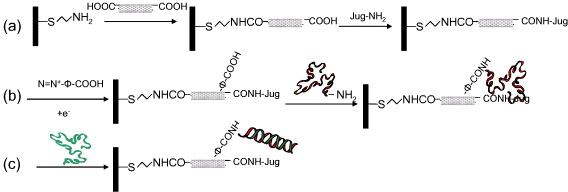
Figure 1 Schematic representation of the successive chemical reactions undergone on the electrode: (a) grafting of cysteamine, SWNT and redox probe; (b) 4-aminobenzoic acid derivatization and grafting of DNA probe; (c) hybridization with complementary DNA target.
5. Experimental
5.1. Chemicals
Cysteamine was provided by Fluka. Dimethylformamide (DMF) was purchased from Sigma; sulfuric acid and nitric acid were from VWR; 4-aminobenzoic acid was supplied by Alfa Aesar; and sodium nitrite from Acros. Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-Hydroxysuccinimide (NHS), single-walled carbon nanotubes (SWNT; dia. 2–10 nm, length 1–5 μm), N,N'-Dicyclohexylcarbodiimide (DCC), 5-hydroxy-1,4-naphthoquinone (Jug), phosphate buffer saline (PBS), lithium perchlorate, 1,3-propanediamine, N-tert- Butoxycarbonyl-2-aminoethanethiol and Au-coated silicon wafers (with 111 surface orientation) were purchased from Aldrich. Au disk electrodes (surface area 0.03 cm 2) were purchased from BASI.
Aqueous solutions were made with Milli-Q water. All oligonucleotide fragments were purchased from Eurogentec and these are described below (bases really used for hybridization are lettered in bold, others are spacers to avoid edge effects, and mismatches are underlined). The probe sequence is GEM-C 6-NH 2 (5'-TCG CAC CCA TCT CTC TCC TTC TAG CCT-3'C 6 H 12 NH 2), the complementary sequence is a fragment tag of the AIDS virus, HIV (3'-CG TGG GTA GAG AGA GGA AGA-5'), the random sequence is RAND (3'-GG TAA ATG ATC CTT CAA CTA-5') and the single mismatched sequence is MHIV (3'-CG TGG GTA AAG AGA GGA AGA-5').
3-[(2-aminoethyl)sulfanyl-5-hydroxy-1,4-naphthoquinone (Jug-NH 2) was synthesized in a two-step reaction. (i) One equivalent of Juglone and one equivalent of N-tert-butoxycarbonyl-2-aminoethanethiol were added in ethanol and stirred for 4 h at room temperature. The solvent was then removed under low pressure. Cyclohexane was added and the solution was allowed to precipitate for 18 h at 5 °C, then filtered and washed with cold ethanol to obtain a dark green powder, which is Jug-NHBoc. (ii) Jug-NHBoc was dissolved in a solution of trifluoroacetic acid in dichloromethane, and stirred 2 h for the deprotection of Boc group. Then all of the solvent and acid were removed under low pressure and the product was deprotonated by adding NaHCO 3. Finally, Jug-NH 2 was obtained by extraction with dichloromethane. HNMR (δH, 200 MHz, CDCl 3, Me 4 Si): 14.5 (1H, s, OH); 7.58 (1H, m, ph-H); 7.44 (1H, m, ph-H); 7.17 (1H, m, ph-H); 6.60 (1H, s, ph-H); 4.32 (2H, m, CH 2) and 2.99 (2H, m, CH 2).
5.2. Electrochemical experiments
Au disks electrodes (area 0.03 cm 2) were purchased from BASI and Au-coated silicon wafers (111) were purchased from Aldrich. For all electrochemical experiments, a conventional one-compartment, three-electrode cell was used with an Au disk as the working electrode, a platinum grid as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The Au wafers used had a surface area of 1 cm 2. Measurements were performed with an Autolab (PGSTAT 30) controlled by GPES software. The grafting of Jug-NH 2 was confirmed by reading the quinone redox couple in PBS using cyclic voltammetry. Hybridization was monitored by following the electroactivity modification of the quinone group using square wave voltammetry (SWV). The following set of parameters was used: pulse height 50 mV, pulse width 50 ms, scan increment 2 mV, frequency 12.5 Hz, and potential range (−0.6; −0.15 V versus SCE).
5.3. SWNT oxidation
10 mg of SWNTs was added to 30 ml of a mixture of sulfuric acid and nitric acid with a 3 : 1 volume ratio. The mixture was sonicated at 60 °C for 4 h, then it was diluted in 1 litre of water. The oxidized SWNTs were obtained by vacuum filtration on a 0.2 μm nitrocellulose membrane (VWR) and washed with water until the filtrate achieved a neutral pH. The oxidized SWNT was dried under vacuum at room temperature for 48 h and characterized by x-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM).
5.4. Generation of a cysteamine self-assembled monolayer on Au
Au disk electrodes were used for electrochemical measurements and Au wafers were used for XPS, AFM (Nanoscope IIIa Digital Instrument; cantilevers: 285 kHz, 42 N m −1; tapping mode) characterizations. Hybridization was detected using fluorescence. Before the surface treatment, Au disk electrodes were polished with 2 alumina slurries of different sizes (1 and 0.3 μm). Then, electrodes and wafers were ultrasonicated in acetone and ethanol and then put inside an UV-Ozone cleaner for 5 min. After the surface pretreatment, they were immersed in s solution of cysteamine (1 mg ml −1) in ethyl acetate at room temperature for 2 h. Finally, they were rinsed thoroughly with ethyl acetate and dried under an argon atmosphere.
5.5. Juglone-modified SWNT immobilization on a cysteamine self-assembled monolayer
A suspension of SWNT-COOH in DMF (0.4 mg ml −1) was prepared and ultrasonicated for 2 h to evenly disperse the SWNTs, then DCC (0.5 mg ml −1) was added and the solution was incubated for 1 h to transform carboxylic acid on SWNTs into an activated ester. Au electrodes or wafers modified with a cysteamine monolayer were immersed in this SWNT solution for 12 h at room temperature. After removal from the solution, they were rinsed in ethanol and allowed to dry.
To attach the redox probe (Jug-NH 2) on free carboxylic acids on SWNTs, the electrodes were immersed into a Jug-NH 2/DMF solution (1 mg ml −1) containing DCC (0.5 mg ml −1) for 7 h at room temperature. They were finally rinsed in ethanol and dried with Ar.
5.6. DNA probe grafting via electrochemical reduction of a diazonium salt
An aqueous solution of aryl diazonium salt was prepared containing 10−3 M 4-aminobenzoic acid, 10−3 M NaNO 2, 10−1 M LiClO 4 and 10−1 M HCl. Jug-SWNT modified Au electrodes and wafers were treated in this solution by chronoamperometry with the following parameters: potential –0.5 V versus SCE, duration 0.5 s. The electroactivity of the system was checked by SWV. After that, the substrates were rinsed in water and immersed in 1 μM ODN (GEM-C 6-NH 2) probe solution containing 10−3 M EDC and 10−3 M NHS for 12 h. Finally, to remove the adsorbed ODN probes, they were immersed in phosphate buffer solution (PBS) for 1 h. The electroactivity change of the quinone groups due to ODN grafting was analyzed by SWV.
5.7. Hybridization protocol and detection
The ODN probe-modified Au electrodes were immersed into PBS (pH 7.4) solution containing 0.1 μM ODN target (complementary, mismatch and random respectively) for 2 h at room temperature. After that, they were immersed in PBS to remove non-hybridized ODN target. The quinone electroactivity after hybridization was analyzed by SWV. The effect of hybridization was reflected by the change in quinone electroactivity.
To ensure that hybridization had really taken place, fluorescence detection was carried out using ODN probe-modified Au wafers. The same hybridization protocol was strictly followed. The only difference was that for Au wafers, all kinds of ODN targets used were modified with fluoresceine on their 5'-end, for which the excitation wavelength is 494 nm and the emission wavelength is 521 nm. After hybridization, a four step washing process was undertaken. For the first 3 steps, the wafers were soaked in fresh PBS for 15 min at room temperature to remove the non-hybridized ODN. In the last step, they were soaked in pure hot water for 10 min at 80 °C. Under these conditions, we assumed that all of the hybridized targets were denatured and put into solution. The solutions after every washing step were collected for fluorescence detection using a spectrophotofluorometer (Fluoromax-4, HORIBA Jobin Yvon). To determine the ODN concentration in these samples, calibration curves were obtained for fluorescent target solutions in the concentration range of 10−8 to 10−10 M. The surface concentrations (Γ, mol cm −2) were calculated using the formula Γ=C×V×A −1, with C being the concentration of target in the washing solution, V the volume of washing solution and A the wafer area.
6. Results and discussion
6.1. SWNT oxidation and immobilization
We first characterized as-received SWNTs and oxidized SWNTs by Transmission Electron Microscopy (TEM) and x-ray Photoelectron Spectroscopy (XPS). TEM images show that SWNTs were cut from more than 1 μm tubes into tubes with lengths of about 50 nm to 500 nm. The XPS C 1s spectrum of oxidized SWNTs (in powder form) shows a novel band at 289 eV, due to the carboxyl moieties generated on the nanotubes (result not shown).
Figure 2 shows the XPS and Atomic Force Microscopy (AFM) characterizations of SWNT grafted on the cysteamine self-assembled monolayer (SAM). In the C 1s spectrum of a pure SAM surface (figure 2(A-1)), 3 peaks are present. The largest peak at 285 eV (C1) was also seen for cleaned gold surfaces and hence stems from usual carbon contamination. The peak at 286.3 eV (C2) is correlated to the mono-layer of cysteamine [36], and a very weak peak above 288 eV (C3) most likely comes from residual solvent (ethyl acetate) on the surface after generation of the mono-layer. After SWNT immobilization (figure 2(A-2)), a significant increase in the oxygen ratio on the surface is observed (not shown) and also an obvious increase in the peak above 288 eV (C3) is detected. This band is certainly due to the carboxylic acid groups carried on SWNTs. AFM images (figure 2(B)) indicate an increase in surface roughness (compare figure 2(B-2) with figure 2(B-1)) caused by the presence of SWNT bundles on the surface. The structure of figure 2(B-2) suggests a vertical alignment of the grafted SWNTs.
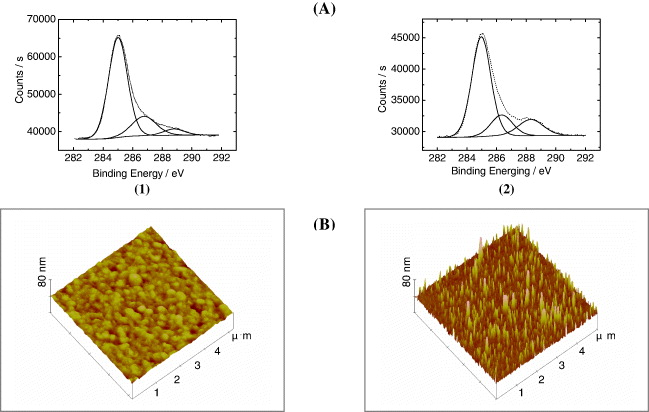
Figure 2 Characterizations of the SWNT immobilization. (A) XPS of Au wafer surface after (1) SAM of cysteamine; after (2) immobilization of SWNTs. (B) AFM images of Au wafer surface after (1) cysteamine grafting; after (2) immobilization of SWNT.
6.2. Electroactivity of Jug-NH2/ SWNT
Once 3-[(2-aminoethyl)sulfanyl-5-hydroxy-1,4-naphthoquinone (Jug-NH 2) was grafted onto the SWNTs, cyclic voltammetry was performed in phosphate buffer solution (PBS). The electroactivity of the quinone end-group is shown in figure 3 (curve b). A quasi-reversible signal is observed with a cathodic and anodic peak at −0.54 and −0.45 V, respectively, for the main couple. The peak currents are a linear function of the scan rate (figure 4). This indicates that the current is limited by the electronic transfer. A shoulder appears at −0.26 V and −0.32 V, that could also be attributed to the quinone electroactivity (juglone dissolved in PBS has the same behavior, one main peak and a shoulder). Integration of the area under the faradic peak gave a charge of about 2.3×10−7 C. This amount corresponds to a Jug surface concentration of 40 pmol cm −2. The response of an electrode modified with a cysteamine SAM+SWCNT layer (figure 3, curve a) appears purely capacitive.
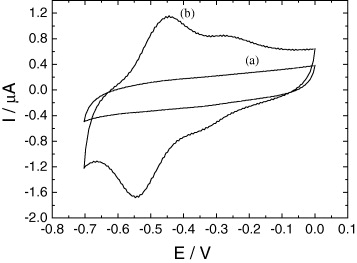
Figure 3 Electroactivity of Jug-NH 2 grafted onto SWNTs (as described in figure 1(a)) in phosphate buffered solution (pH 7.4). Electrode surface 0.03 cm 2. v=100 mV ·s −1. (a) Cyclic voltammogram of a gold electrode after grafting of cysteamine + SWCNTs onto it; (b) cyclic voltammogram of the same electrode after the grafting of Jug-NH 2 onto SWNTs.
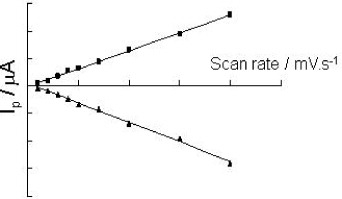
Figure 4 Evolution of the cathodic (▪) and anodic (▴) faradic peak currents with the scan rate for an electrode modified by SWNTs and Jug-NH 2 (same conditions as figure 3).
6.3. ODN probe grafting on SWNTs
For ODN probe grafting, new carboxylic acid sites were introduced on the SWNTs by reduction of the diazonium salt of 4-amino benzoic acid, as described in the experimental section, with a short reduction time of 0.5 s. After that, the change in electroactivity of juglone was followed by SWV in PBS. We found a significant current decrease (30%) compared with the signal before the diazonium treatment. With such a short reduction time, it is difficult to form a layer that can block the electrode surface. So this current decrease could be explained by a change in SWCNT conductivity after diazonium grafting on the walls of the SWCNTs.
The system ODN probe/Jug-NH 2/SWNT was characterized by square wave voltammetry (SWV) from −0.6 V to −0.15 V versus SCE in PBS (figure 5). A current decrease of about 40% was detected compared to the Jug-NH 2/SWNT system. This behavior is attributed to the fact that the ODN probe strand generates hydrogen bonding with Jug-NH 2, between bases from ODN and quinone from Jug-NH 2 [37–40].
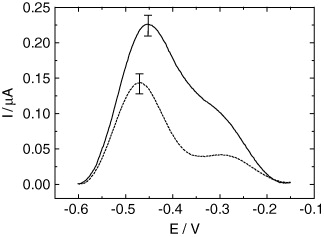
Figure 5 SWV responses obtained for the Au electrode modified by cysteamine, SWNTs, Jug-NH 2 and 4-aminobenzoic acid before ODN-probe grafting (a) and after ODN-probe grafting (b). Medium: PBS. Electrode surface: 0.03 cm 2.
6.4. Fluorescent hybridization detection
Fluorescent detection for a hybridization event is a traditional and reliable technique. That is why we used this approach, as described in the experimental section. As shown in figure 6, less than 1 pmol cm −2 fluorescent targets were removed after the first 3 washing steps for every sample. This quantity corresponds to non-specific physisorption of the target strands, as it is independent of the sequence. In contrast, the stringent conditions (80 °C, pure H 2 O) of the fourth step result in the denaturation of all hybridized ODN. We can see that this step leads to dehybridization of approximately 1.5 pmol cm −2 for the complementary strand (black bar) but a negligible amount for random and mismatch strands (hatch bars). These results show that hybridization has taken place selectively only with the complementary target.
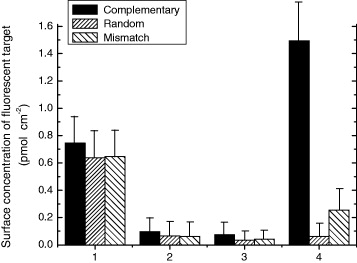
Figure 6 Surface concentration of fluorescent targets obtained after three washing steps (1–3) and dehybridization (4) (see experimental section for conditions).
6.5. Electrochemical hybridization detection
As mentioned above, the influence of a single-stranded ODN on the juglone moiety will change its electroactivity, due to the steric effect and hydrogen interactions between the two [37–40]. These interactions are expected to decrease after hybridization with the complementary target, because the more rigid double strand that forms takes less space and bases involved in Watson–Crick pairing are no longer available to form hydrogen bonds with juglone. Figure 7 shows the differential peak current (see figure caption for details) of SWV analysis before and after incubation with different targets (complementary, random and single mismatch). It appears that the complementary target (HIV) leads to an obvious increase in the current. In contrast, for random and mismatch targets, the current change is not significant. These results corroborate data from fluorescence detection and support our assumptions. The system thus shows good selectivity as it allows discrimination between complementary (HIV) and random (RAND) sequences, as well as between sequences that have just one base changed (HIV and MHIV). With the complementary sequence, the current response gains about +85% of its initial value after hybridization, compared to about +35% for previous results obtained with a conducting polymer architecture, of a self-assembled alkylthiol architecture [37].
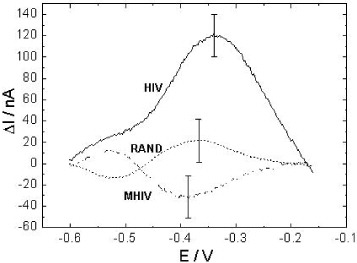
Figure 7 Differential peak current (ΔI=I HYB −I GEM ), with I HYB and I GEM as the peak currents after and before hybridization, respectively, for electrodes modified by cysteamine, SWNTs, Jug-NH 2 and GEM probe, after incubation with the complementary sequence (HIV), the random sequence (RAND) and the mismatch sequence (MHIV) (conditions detailed in the experimental section).
7. Conclusions
In conclusion, a label-free, reagentless electrochemical DNA hybridization sensing strategy was developed using SWNTs as nanoparticles to bring 2 different probes (DNA and redox probes put close together). Shortened SWNTs were covalently and vertically immobilized on a self-assembled monolayer of cysteamine. Then a DNA probe and a redox probe were respectively grafted onto SWNTs. In this way, the distribution of the DNA and redox probes over the electrode surface could be controlled by nanostructuration induced by SWNTs. This improved non-covalent interactions between them, so that redox current variations induced by the conformational change of the DNA probe could be improved as well. For further developments, it would be useful to consider SWNTs of more homogenous physical properties (i.e. length, diameter and conductivity), which may become easier to obtain with the development of CNT fabrication and processing. In addition, non-covalent functionalization of SWNTs could be envisaged, e.g. by functionalization of the redox and DNA probes with pyrene.
Acknowledgments
QDZ thanks the French Ministry of Research for a PhD grant and University Paris Diderot—Paris 7 for a teaching position.